Member Spotlight
Dr. Edward Hubbard is a faculty member in Educational Psychology within the School of Education. Dr. Hubbard runs the Educational Neuroscience Lab at UW-Madison and is active in research, particularly regarding structures and properties of the brain to develop and improve educational practices. He started as a member of the ED/SBS IRB in 2016 and transitioned to Vice-Chair of the committee in 2018. Shortly after the ED/SBS IRB merged with the Minimal Risk IRB in June 2021 to form the new Minimal Risk Research (MRR) IRB, he assumed the role of Chair of the MRR IRB. His research crosses both realms of clinical and educational research, which makes him the perfect Chair for the committee. Dr. Hubbard also brings years of IRB experience to his role as Chair of the MRR IRB. Thank you for your service to the IRB, Dr. Hubbard!
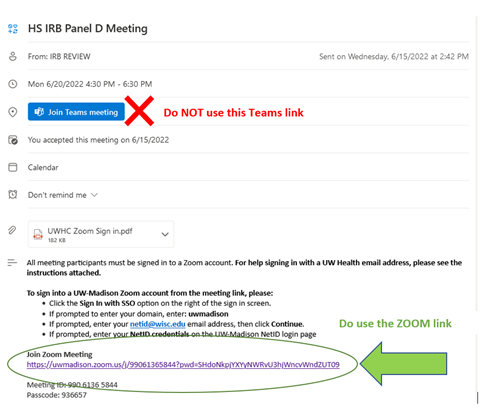
Reminder: IRB Meetings will Always use Zoom
You may have noticed that Outlook calendar invites for IRB meetings include both a Teams link as well as a Zoom link. Unfortunately, the Teams link is automatically added by Outlook with each invite and this is leading to much confusion. IRB meetings will ALWAYS USE ZOOM. Please ignore the Teams link. This issue will be corrected with the 2023 meeting invitations.
For now, if you log into Teams, you will see a message directing you to the Zoom link.
AAHRPP Site Visit
As most of you know, we recently had our AAHRPP reaccreditation site visit. As part of this process, many of our MRR and HS members and Chairs/Vice-Chairs were interviewed. We would like to thank each of you who participated in this process for taking the time to prepare and sit for these interviews.
The AAHRPP site visitors had some wonderful things to say about our program, specifically our pre-submission IRB Consultation services, the RELIANT Team, and the FDA/IND/IDE resources we have here on campus. They also reported that our researchers and research staff appreciate the work the work we are doing to make our IRB review processes as efficient and effective as possible, praised the helpfulness of the IRB staff, and view the IRB as a resource and not an obstacle.
There are a few niche areas of concern that we are already working on so that a reply is ready when we receive our Site Visit Report.
Our dedicated IRB members are a critical part of our HRPP. THANK YOU to each of you for the important role that you play in maintaining our high standards.
July 1 Evaluation Cycle
Federal regulations as well as AAHRP accreditation standards require that IRB members be evaluated annually. Although for many of you, it will feel like we just completed this process, we are working towards getting all of our new and experienced members on the same annual evaluation cycle. To this end, we are beginning this process for the July 1 2021-June 30 2022 evaluation period. Going forward, this will allow us to complete these for all members coinciding with the fiscal year, regardless of when they became a member.
As a reminder, evaluations include both a quantitative and qualitative portion. The quantitative portion includes things like the number of meetings attended and the number of reviews assigned. This helps to give the IRB office a sense of the time commitment required. The qualitative portion assesses things like preparedness and contributions at meetings. We of course know that we have many new members who are still learning the complexities of the IRB review process, so this also gives us an opportunity to identify future training needs and is meant to be a transparent and collaborative process. The evaluation tool is in the Toolkit Library on our website and available for your review.
You will receive communications from the IRB office mid to late September about your evaluation. Members are encouraged to reach out to their panel administer or chair with any feedback on the evaluation and will be given the opportunity to provide more formal feedback in the Member Satisfaction Survey which will go out along with the evaluations.
Continuing Education: Waivers of consent and Waivers of Documentation of Consent
Waiver of the Requirement to Obtain Informed Consent
An IRB may waive the requirement to obtain consent if it finds that all of the following conditions are met:
- The research involves no more than minimal risk to the subjects;
- The waiver will not adversely affect the rights and welfare of the subjects;
- If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format;
- The research could not practicably be carried out without the waiver;
- Whenever appropriate, the subjects or LARs will be provided with additional pertinent information after participation.
Waivers of consent are most typically granted in cases where research involves activities that will not involve interactions with subjects and are limited to the collection of data (e.g., clinical records) or specimens that have, or will, come into existence as part of non-research-related clinical activities. A frequently challenging consideration is determining whether the research would be impracticable without the waiver. Considerations favoring an impracticability determination usually involve expected difficulties in contacting and/or obtaining responses from subjects (e.g., due to the number of subjects, availability of contact information, resource requirements, etc.). Determining whether a waiver would adversely affect the rights and welfare of subjects can also raise difficulties. In cases where a waiver of consent may be expected to be contrary to subject preferences (e.g., highly sensitive genetic testing, creation of cell lines, study results that could be stigmatizing to ethnic groups, etc.), there may be concerns that that this would be contrary to subject rights and welfare.
In rare cases, a waiver of consent could be sought to allow for the enrollment of minors in research without parent or guardian consent. Such cases would typically require subjects who are capable of providing meaningful assent and involve research where parent/guardian involvement would not be possible or would place subjects at risk (e.g., population of homeless youth, subjects engaged in illicit activities, etc.).
Waiver of Requirement to Obtain Written Informed Consent
An IRB may waive the requirement to obtain a signed consent form if it finds that ANY of the following conditions are met:
- That the only record linking the subject and the research would be the informed consent form and the principal risk would be potential harm resulting from a breach of confidentiality. Each subject (or LAR) will be asked whether the subject wants documentation linking the subject with the research, and the subject’s wishes will govern;
- That the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context; or
- If the subjects or LARs are members of a distinct cultural group or community in which signing forms is not the norm, that the research presents no more than minimal risk of harm to subjects and provided there is an appropriate alternative mechanism for documenting that informed consent was obtained.
Waivers of signed consent are most typically granted under category 2, where research participation involves minimal risk activities and consent procedures will not occur in person. Common examples include the use of an oral consent script or consent document emailed to subjects prior to a phone or virtual consent discussion. Subjects would provide oral consent and researchers would document the receipt of the latter. In addition, online questionnaire studies frequently involve a waiver of signed consent. In such cases, an introductory consent document is typically incorporated into the instrument, and it is specified that completion of the instrument constitutes consent.